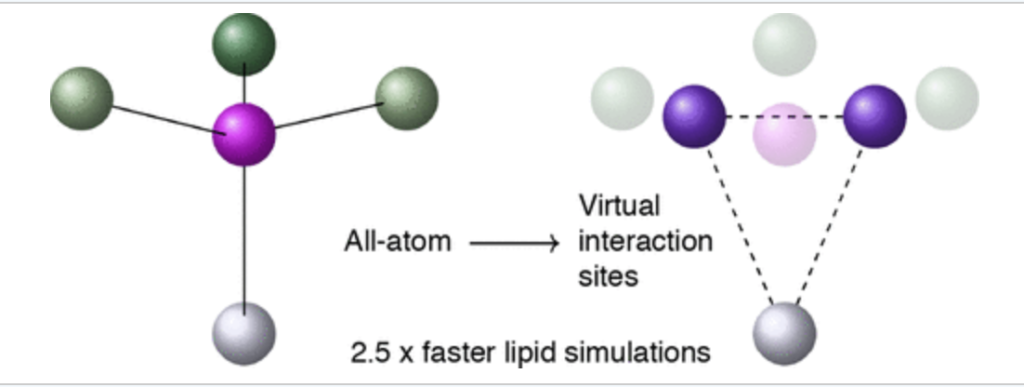
We have implemented the virtual sites technique for lipid membranes, which completely suppresses the degrees of freedom of the hydrogen atoms in a lipid bilayer allowing for an increased time step of 5 fs in all-atom simulations of the CHARMM36 force field. The hydrogen virtual sites still have non-bonded interactions, and thus the method is more “accurate” than the united force field method, where information on hydrogen atoms is lost completely.
The method performs well for nearly all standard lipid types. We are currently working on application of the method to other force fields and lipids.
The positions of the H atoms on the tertiary amino group of a lipid headgroup can be calculated from the positions of two virtual site particles.